

All organic operate depends on how completely different proteins work together with one another. Protein-protein interactions facilitate all the things from transcribing DNA and controlling cell division to higher-level capabilities in complicated organisms.
A lot stays unclear, nevertheless, about how these capabilities are orchestrated on the molecular stage, and the way proteins work together with one another — both with different proteins or with copies of themselves.
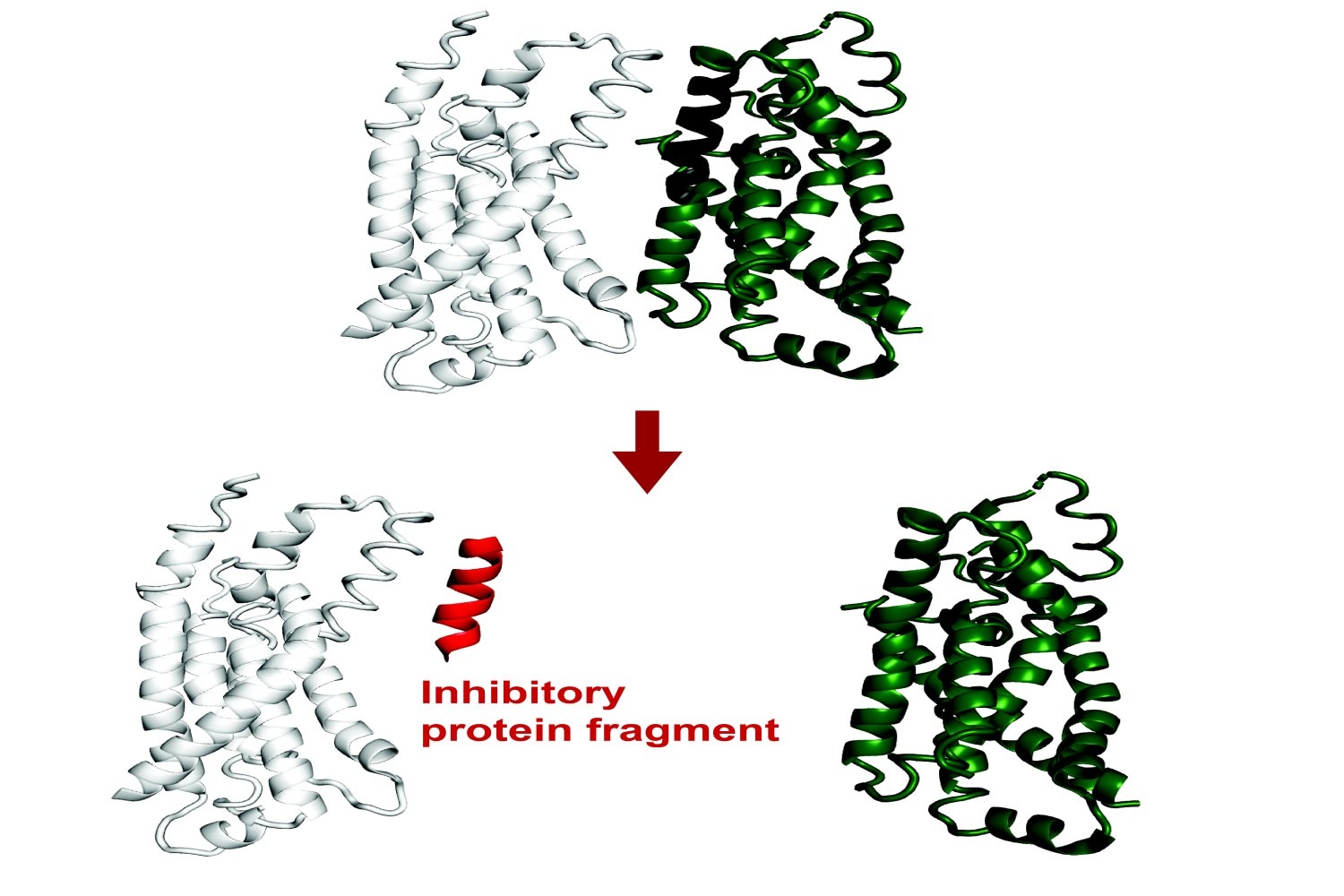
Current findings have revealed that small protein fragments have a number of practical potential. Despite the fact that they’re incomplete items, brief stretches of amino acids can nonetheless bind to interfaces of a goal protein, recapitulating native interactions. By means of this course of, they will alter that protein’s operate or disrupt its interactions with different proteins.
Protein fragments might subsequently empower each primary analysis on protein interactions and mobile processes, and will doubtlessly have therapeutic functions.
Just lately revealed in Proceedings of the Nationwide Academy of Sciences, a brand new methodology developed within the Division of Biology builds on current synthetic intelligence fashions to computationally predict protein fragments that may bind to and inhibit full-length proteins in E. coli. Theoretically, this instrument might result in genetically encodable inhibitors towards any protein.
The work was completed within the lab of affiliate professor of biology and Howard Hughes Medical Institute investigator Gene-Wei Li in collaboration with the lab of Jay A. Stein (1968) Professor of Biology, professor of organic engineering, and division head Amy Keating.
Leveraging machine studying
This system, known as FragFold, leverages AlphaFold, an AI mannequin that has led to phenomenal developments in biology lately as a result of its means to foretell protein folding and protein interactions.
The objective of the challenge was to foretell fragment inhibitors, which is a novel utility of AlphaFold. The researchers on this challenge confirmed experimentally that greater than half of FragFold’s predictions for binding or inhibition have been correct, even when researchers had no earlier structural information on the mechanisms of these interactions.
“Our outcomes recommend that it is a generalizable method to seek out binding modes which are more likely to inhibit protein operate, together with for novel protein targets, and you should use these predictions as a place to begin for additional experiments,” says co-first and corresponding writer Andrew Savinov, a postdoc within the Li Lab. “We are able to actually apply this to proteins with out recognized capabilities, with out recognized interactions, with out even recognized buildings, and we are able to put some credence in these fashions we’re growing.”
One instance is FtsZ, a protein that’s key for cell division. It’s well-studied however incorporates a area that’s intrinsically disordered and, subsequently, particularly difficult to review. Disordered proteins are dynamic, and their practical interactions are very possible fleeting — occurring so briefly that present structural biology instruments can’t seize a single construction or interplay.
The researchers leveraged FragFold to discover the exercise of fragments of FtsZ, together with fragments of the intrinsically disordered area, to determine a number of new binding interactions with varied proteins. This leap in understanding confirms and expands upon earlier experiments measuring FtsZ’s organic exercise.
This progress is critical partially as a result of it was made with out fixing the disordered area’s construction, and since it reveals the potential energy of FragFold.
“That is one instance of how AlphaFold is basically altering how we are able to research molecular and cell biology,” Keating says. “Artistic functions of AI strategies, corresponding to our work on FragFold, open up surprising capabilities and new analysis instructions.”
Inhibition, and past
The researchers achieved these predictions by computationally fragmenting every protein after which modeling how these fragments would bind to interplay companions they thought have been related.
They in contrast the maps of predicted binding throughout your complete sequence to the results of those self same fragments in residing cells, decided utilizing high-throughput experimental measurements through which hundreds of thousands of cells every produce one sort of protein fragment.
AlphaFold makes use of co-evolutionary info to foretell folding, and sometimes evaluates the evolutionary historical past of proteins utilizing one thing known as a number of sequence alignments for each single prediction run. The MSAs are crucial, however are a bottleneck for large-scale predictions — they will take a prohibitive period of time and computational energy.
For FragFold, the researchers as an alternative pre-calculated the MSA for a full-length protein as soon as, and used that outcome to information the predictions for every fragment of that full-length protein.
Savinov, along with Keating Lab alumnus Sebastian Swanson PhD ’23, predicted inhibitory fragments of a various set of proteins along with FtsZ. Among the many interactions they explored was a posh between lipopolysaccharide transport proteins LptF and LptG. A protein fragment of LptG inhibited this interplay, presumably disrupting the supply of lipopolysaccharide, which is a vital part of the E. coli outer cell membrane important for mobile health.
“The massive shock was that we are able to predict binding with such excessive accuracy and, in truth, typically predict binding that corresponds to inhibition,” Savinov says. “For each protein we’ve checked out, we’ve been capable of finding inhibitors.”
The researchers initially targeted on protein fragments as inhibitors as a result of whether or not a fraction might block a vital operate in cells is a comparatively easy final result to measure systematically. Trying ahead, Savinov can be eager about exploring fragment operate exterior inhibition, corresponding to fragments that may stabilize the protein they bind to, improve or alter its operate, or set off protein degradation.
Design, in precept
This analysis is a place to begin for growing a systemic understanding of mobile design rules, and what parts deep-learning fashions could also be drawing on to make correct predictions.
“There’s a broader, further-reaching objective that we’re constructing in direction of,” Savinov says. “Now that we are able to predict them, can we use the info we’ve got from predictions and experiments to drag out the salient options to determine what AlphaFold has truly realized about what makes a superb inhibitor?”
Savinov and collaborators additionally delved additional into how protein fragments bind, exploring different protein interactions and mutating particular residues to see how these interactions change how the fragment interacts with its goal.
Experimentally analyzing the conduct of 1000’s of mutated fragments inside cells, an method often called deep mutational scanning, revealed key amino acids which are accountable for inhibition. In some circumstances, the mutated fragments have been much more potent inhibitors than their pure, full-length sequences.
“In contrast to earlier strategies, we aren’t restricted to figuring out fragments in experimental structural information,” says Swanson. “The core power of this work is the interaction between high-throughput experimental inhibition information and the anticipated structural fashions: the experimental information guides us in direction of the fragments which are significantly attention-grabbing, whereas the structural fashions predicted by FragFold present a selected, testable speculation for a way the fragments operate on a molecular stage.”
Savinov is happy about the way forward for this method and its myriad functions.
“By creating compact, genetically encodable binders, FragFold opens a variety of prospects to control protein operate,” Li agrees. “We are able to think about delivering functionalized fragments that may modify native proteins, change their subcellular localization, and even reprogram them to create new instruments for learning cell biology and treating ailments.”
All organic operate depends on how completely different proteins work together with one another. Protein-protein interactions facilitate all the things from transcribing DNA and controlling cell division to higher-level capabilities in complicated organisms.
A lot stays unclear, nevertheless, about how these capabilities are orchestrated on the molecular stage, and the way proteins work together with one another — both with different proteins or with copies of themselves.
Current findings have revealed that small protein fragments have a number of practical potential. Despite the fact that they’re incomplete items, brief stretches of amino acids can nonetheless bind to interfaces of a goal protein, recapitulating native interactions. By means of this course of, they will alter that protein’s operate or disrupt its interactions with different proteins.
Protein fragments might subsequently empower each primary analysis on protein interactions and mobile processes, and will doubtlessly have therapeutic functions.
Just lately revealed in Proceedings of the Nationwide Academy of Sciences, a brand new methodology developed within the Division of Biology builds on current synthetic intelligence fashions to computationally predict protein fragments that may bind to and inhibit full-length proteins in E. coli. Theoretically, this instrument might result in genetically encodable inhibitors towards any protein.
The work was completed within the lab of affiliate professor of biology and Howard Hughes Medical Institute investigator Gene-Wei Li in collaboration with the lab of Jay A. Stein (1968) Professor of Biology, professor of organic engineering, and division head Amy Keating.
Leveraging machine studying
This system, known as FragFold, leverages AlphaFold, an AI mannequin that has led to phenomenal developments in biology lately as a result of its means to foretell protein folding and protein interactions.
The objective of the challenge was to foretell fragment inhibitors, which is a novel utility of AlphaFold. The researchers on this challenge confirmed experimentally that greater than half of FragFold’s predictions for binding or inhibition have been correct, even when researchers had no earlier structural information on the mechanisms of these interactions.
“Our outcomes recommend that it is a generalizable method to seek out binding modes which are more likely to inhibit protein operate, together with for novel protein targets, and you should use these predictions as a place to begin for additional experiments,” says co-first and corresponding writer Andrew Savinov, a postdoc within the Li Lab. “We are able to actually apply this to proteins with out recognized capabilities, with out recognized interactions, with out even recognized buildings, and we are able to put some credence in these fashions we’re growing.”
One instance is FtsZ, a protein that’s key for cell division. It’s well-studied however incorporates a area that’s intrinsically disordered and, subsequently, particularly difficult to review. Disordered proteins are dynamic, and their practical interactions are very possible fleeting — occurring so briefly that present structural biology instruments can’t seize a single construction or interplay.
The researchers leveraged FragFold to discover the exercise of fragments of FtsZ, together with fragments of the intrinsically disordered area, to determine a number of new binding interactions with varied proteins. This leap in understanding confirms and expands upon earlier experiments measuring FtsZ’s organic exercise.
This progress is critical partially as a result of it was made with out fixing the disordered area’s construction, and since it reveals the potential energy of FragFold.
“That is one instance of how AlphaFold is basically altering how we are able to research molecular and cell biology,” Keating says. “Artistic functions of AI strategies, corresponding to our work on FragFold, open up surprising capabilities and new analysis instructions.”
Inhibition, and past
The researchers achieved these predictions by computationally fragmenting every protein after which modeling how these fragments would bind to interplay companions they thought have been related.
They in contrast the maps of predicted binding throughout your complete sequence to the results of those self same fragments in residing cells, decided utilizing high-throughput experimental measurements through which hundreds of thousands of cells every produce one sort of protein fragment.
AlphaFold makes use of co-evolutionary info to foretell folding, and sometimes evaluates the evolutionary historical past of proteins utilizing one thing known as a number of sequence alignments for each single prediction run. The MSAs are crucial, however are a bottleneck for large-scale predictions — they will take a prohibitive period of time and computational energy.
For FragFold, the researchers as an alternative pre-calculated the MSA for a full-length protein as soon as, and used that outcome to information the predictions for every fragment of that full-length protein.
Savinov, along with Keating Lab alumnus Sebastian Swanson PhD ’23, predicted inhibitory fragments of a various set of proteins along with FtsZ. Among the many interactions they explored was a posh between lipopolysaccharide transport proteins LptF and LptG. A protein fragment of LptG inhibited this interplay, presumably disrupting the supply of lipopolysaccharide, which is a vital part of the E. coli outer cell membrane important for mobile health.
“The massive shock was that we are able to predict binding with such excessive accuracy and, in truth, typically predict binding that corresponds to inhibition,” Savinov says. “For each protein we’ve checked out, we’ve been capable of finding inhibitors.”
The researchers initially targeted on protein fragments as inhibitors as a result of whether or not a fraction might block a vital operate in cells is a comparatively easy final result to measure systematically. Trying ahead, Savinov can be eager about exploring fragment operate exterior inhibition, corresponding to fragments that may stabilize the protein they bind to, improve or alter its operate, or set off protein degradation.
Design, in precept
This analysis is a place to begin for growing a systemic understanding of mobile design rules, and what parts deep-learning fashions could also be drawing on to make correct predictions.
“There’s a broader, further-reaching objective that we’re constructing in direction of,” Savinov says. “Now that we are able to predict them, can we use the info we’ve got from predictions and experiments to drag out the salient options to determine what AlphaFold has truly realized about what makes a superb inhibitor?”
Savinov and collaborators additionally delved additional into how protein fragments bind, exploring different protein interactions and mutating particular residues to see how these interactions change how the fragment interacts with its goal.
Experimentally analyzing the conduct of 1000’s of mutated fragments inside cells, an method often called deep mutational scanning, revealed key amino acids which are accountable for inhibition. In some circumstances, the mutated fragments have been much more potent inhibitors than their pure, full-length sequences.
“In contrast to earlier strategies, we aren’t restricted to figuring out fragments in experimental structural information,” says Swanson. “The core power of this work is the interaction between high-throughput experimental inhibition information and the anticipated structural fashions: the experimental information guides us in direction of the fragments which are significantly attention-grabbing, whereas the structural fashions predicted by FragFold present a selected, testable speculation for a way the fragments operate on a molecular stage.”
Savinov is happy about the way forward for this method and its myriad functions.
“By creating compact, genetically encodable binders, FragFold opens a variety of prospects to control protein operate,” Li agrees. “We are able to think about delivering functionalized fragments that may modify native proteins, change their subcellular localization, and even reprogram them to create new instruments for learning cell biology and treating ailments.”


{kind=link}